AGGRASTAT
-
tirofiban hydrochloride injection, solution
Medicure International Inc
AGGRASTAT¹ (tirofiban hydrochloride), a non-peptide antagonist of the platelet glycoprotein (GP) IIb/IIIa receptor, inhibits platelet aggregation.
Tirofiban hydrochloride monohydrate, a non-peptide molecule, is chemically described as N-(butylsulfonyl)-O-[4-(4-piperidinyl)butyl]-L-tyrosine monohydrochloride monohydrate.
Its molecular formula is C22H36N2O5S•HCl•H2O and its structural formula is:
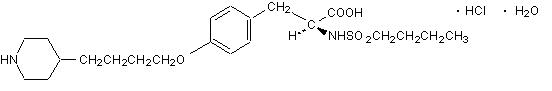
Tirofiban hydrochloride monohydrate is a white to off-white, non-hygroscopic, free-flowing powder, with a molecular weight of 495.08. It is very slightly soluble in water.
AGGRASTAT Injection Premixed is supplied as a sterile solution in water for injection, for intravenous use only, in plastic containers of 100 mL or 250 mL. Each 100 mL of the premixed, iso-osmotic intravenous injection contains 5.618 mg tirofiban hydrochloride monohydrate equivalent to 5 mg tirofiban (50 mcg/mL) and the following inactive ingredients: 0.9 g sodium chloride, 54 mg sodium citrate dihydrate, and 3.2 mg citric acid anhydrous. Each 250 mL of the premixed, iso-osmotic intravenous injection contains 14.045 mg tirofiban hydrochloride monohydrate equivalent to 12.5 mg tirofiban (50 mcg/mL) and the following inactive ingredients: 2.25 g sodium chloride, 135 mg sodium citrate dihydrate, and 8 mg citric acid anhydrous.
The pH of the solution ranges from 5.5 to 6.5 and may have been adjusted with hydrochloric acid and/or sodium hydroxide. The flexible container is manufactured from a specially designed multilayer plastic (PL 2408). Solutions in contact with the plastic container leach out certain chemical components from the plastic in very small amounts; however, biological testing was supportive of the safety of the plastic container materials.
AGGRASTAT is a reversible antagonist of fibrinogen binding to the GP lIb/lIla receptor, the major platelet surface receptor involved in platelet aggregation. When administered intravenously, AGGRASTAT inhibits ex vivo platelet aggregation in a dose- and concentration-dependent manner.
When given according to the recommended regimen, >90% inhibition is attained by the end of the 30-minute infusion. Platelet aggregation inhibition is reversible following cessation of the infusion of AGGRASTAT.
Tirofiban has a half-life of approximately 2 hours. It is cleared from the plasma largely by renal excretion, with about 65% of an administered dose appearing in urine and about 25% in feces, both largely as unchanged tirofiban. Metabolism appears to be limited.
Tirofiban is not highly bound to plasma proteins and protein binding is concentration independent over the range of 0.01 to 25 mcg/mL. Unbound fraction in human plasma is 35%. The steady state volume of distribution of tirofiban ranges from 22 to 42 liters.
In healthy subjects, the plasma clearance of tirofiban ranges from 213 to 314 mL/min. Renal clearance accounts for 39 to 69% of plasma clearance. The recommended regimen of a loading infusion followed by a maintenance infusion produces a peak tirofiban plasma concentration that is similar to the steady state concentration during the infusion. In patients with coronary artery disease, the plasma clearance of tirofiban ranges from 152 to 267 mL/min; renal clearance accounts for 39% of plasma clearance.
Plasma clearance of tirofiban in patients with coronary artery disease is similar in males and females.
Plasma clearance of tirofiban is about 19 to 26% lower in elderly (>65 years) patients with coronary artery disease than in younger (≤65 years) patients.
No difference in plasma clearance was detected in patients of different races.
In patients with mild to moderate hepatic insufficiency, plasma clearance of tirofiban is not significantly different from clearance in healthy subjects.
Plasma clearance of tirofiban is significantly decreased (>50%) in patients with creatinine clearance <30 mL/min, including patients requiring hemodialysis (see DOSAGE AND ADMINISTRATION, Recommended Dosage).Tirofiban is removed by hemodialysis.
AGGRASTAT inhibits platelet function, as demonstrated by its ability to inhibit ex vivo adenosine phosphate (ADP)-induced platelet aggregation and prolong bleeding time in healthy subjects and patients with coronary artery disease. The time course of inhibition parallels the plasma concentration profile of the drug.
Following discontinuation of an infusion of AGGRASTAT, 0.10 mcg/kg/min, ex vivo platelet aggregation returns to near baseline in approximately 90% of patients with coronary artery disease in 4 to 8 hours. The addition of heparin to this regimen does not significantly alter the percentage of subjects with >70% inhibition of platelet aggregation (IPA), but does increase the average bleeding time, as well as the number of patients with bleeding times prolonged to >30 minutes.
In patients with unstable angina, a two-staged intravenous infusion regimen of AGGRASTAT (loading infusion of 0.4 mcg/kg/min for 30 minutes followed by 0.1 mcg/kg/min for up to 48 hours in the presence of heparin and aspirin), produces approximately 90% inhibition of ex vivo ADP-induced platelet aggregation with a 2.9-fold prolongation of bleeding time during the loading infusion. Inhibition persists over the duration of the maintenance infusion.
Three large-scale clinical studies were conducted to study the efficacy and safety of AGGRASTAT in the management of patients with Acute Coronary Syndrome (unstable angina/non-Q-wave myocardial infarction). Acute Coronary Syndrome is characterized by prolonged (≥10 minutes) or repetitive symptoms of cardiac ischemia occurring at rest or with minimal exertion, associated with either ischemic ST-T wave changes on electrocardiogram (ECG) or elevated cardiac enzymes. The definition includes “unstable angina” and “non-Q-wave myocardial infarction” but excludes myocardial infarction that is associated with Q-waves or non-transient ST-segment elevation. The three studies examined AGGRASTAT alone and as an addition to heparin, prior to and after angioplasty (if indicated) (PRISM-PLUS), in comparison to heparin in a similar population (PRISM), and in addition to heparin in patients undergoing percutaneous transluminal coronary angioplasty (PTCA) or atherectomy (RESTORE). These trials are discussed in detail below.
In the multi-center, randomized, parallel, double-blind PRISM-PLUS trial, the use of AGGRASTAT in combination with heparin (n=773) was compared to heparin alone (n=797) in patients with documented unstable angina/non-Q-wave myocardial infarction within 12 hours of entry into the study and initiation of treatment. All patients with unstable angina/non-Q-wave myocardial infarction had cardiac ischemia documented by ECG or had elevated cardiac enzymes. Patients who were medically managed or who subsequently underwent revascularization procedures were studied. The mean age of the population was 63 years; 32% of patients were female and approximately half of the population presented with non- Q-wave myocardial infarction. Exclusions included contraindications to anticoagulation (see CONTRAINDICATIONS), decompensated heart failure, platelet count <150,000/mm3, and creatinine >2.5 mg/dL. In this study, patients were randomized to either AGGRASTAT (30 minute loading infusion of 0.4 mcg/kg/min followed by a maintenance infusion of 0.10 mcg/kg/min) and heparin (bolus of 5,000 units (U) followed by an infusion of 1,000 U/hr titrated to maintain an activated partial thromboplastin time (APTT) of approximately 2 times control), or heparin alone (bolus of 5,000 U followed by an infusion of 1,000 U/hr titrated to maintain an APTT of approximately 2 times control). All patients received concomitant aspirin unless contraindicated. Patients underwent 48 hours of medical stabilization on study drug therapy, and they were to undergo angiography before 96 hours (and, if indicated, angioplasty/atherectomy, while continuing on AGGRASTAT and heparin for 12-24 hours after the procedure). Some patients went on to coronary artery bypass grafting (CABG) after cessation of drug therapy. AGGRASTAT and heparin could be continued for up to 108 hours. On average, patients received AGGRASTAT for 71.3 hours. A third group of patients was initially randomized to AGGRASTAT alone (no heparin). This arm was stopped when the group was found, at an interim look, to have greater mortality than the other two groups. Note, however, that a direct comparison of heparin and tirofiban alone in the PRISM study (see below) did not show excess mortality.
The primary endpoint of the study was a composite of refractory ischemia, new myocardial infarction and death at 7 days after initiation of AGGRASTAT and heparin. At the primary endpoint, there was a 32% risk reduction in the overall composite. The components of the composite were examined separately (they total more than the composite because a patient could have more than one, e.g., by dying after having a new infarction). There was a 47% risk reduction in myocardial infarction and a 30% risk reduction in refractory ischemia. The results are shown in Table 1.
| Endpoint | AGGRASTAT+Heparin (n=773) |
Heparin (n=797) |
Risk Reduction | p-value |
| Composite Endpoint | 12.9% | 17.9% | 32% | 0.004 |
| Components | ||||
| Myocardial Infarction and Death | 4.9% | 8.3% | 43% | 0.006 |
| Myocardial Infarction | 3.9% | 7.0% | 47% | 0.006 |
| Death | 1.9% | 1.9% | — | — |
| Refractory Ischemia | 9.3% | 12.7% | 30% | 0.023 |
The benefit seen at 7 days was maintained over time. At 30 days, the risk of the composite endpoint was reduced by 22% (p=0.029) and there was a 30% reduction in the composite of myocardial infarction and death (p=0.027). At 6 months, the risk of the composite endpoint was reduced by 19% (p=0.024). The risk reduction in the composite endpoint at 30 days and 6 months is shown in the Kaplan-Meier curve below.
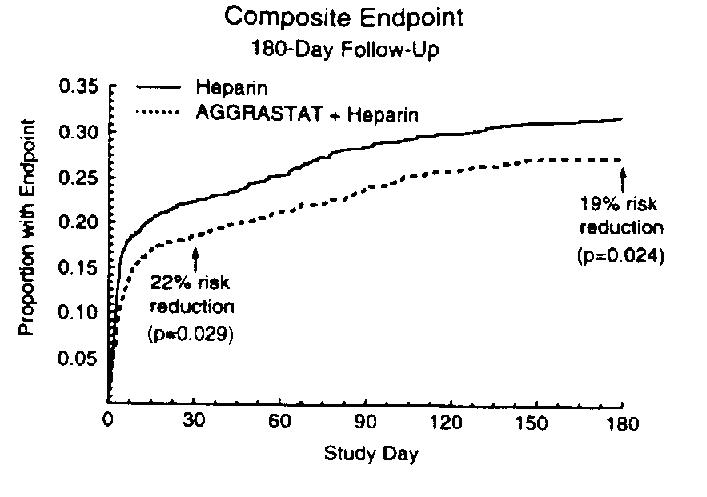
PRISM-PLUS was not designed to provide definitive results in subsets of the overall population. Nonetheless, results were examined for demographic (age, gender, race) subsets and for people who did and did not receive PTCA, atherectomy, or CABG.
In PRISM-PLUS, there was a consistent treatment effect in patients either greater or less than 65 years old. Too few non-Caucasians were enrolled to make a definite statement about racial differences in treatment effect.
An analysis of the results by sex suggests that women who are medically managed or who undergo subsequent percutaneous transluminal coronary angioplasty/atherectomy may receive less benefit from tirofiban (95% confidence limits for relative risk of 0.61-1.74) than do men (0.43-0.89) (p=0.11). This difference may be a true treatment difference, the effect of other differences in these subgroups, or a chance occurrence.
Approximately 90% of patients in the PRISM-PLUS study underwent coronary angiography and 30% underwent angioplasty/atherectomy during the first 30 days of the study. The majority of these patients continued on study drug throughout these procedures. AGGRASTAT was continued for 12-24 hours (average 15 hours) after angioplasty/atherectomy. The effects of AGGRASTAT at Day 30 did not appear to differ among the sub-populations that did or did not receive PTCA or CABG, both prior to and after the procedure.
A sub-study in PRISM-PLUS of angiograms after 48 to 96 hours found that there was a significant decrease in the extent of angiographically apparent thrombus in patients treated with AGGRASTAT in combination with heparin compared to heparin alone. In addition, flow in the affected coronary artery was significantly improved.
In the PRISM study, a randomized, parallel, double-blind, active control study, AGGRASTAT alone (n=1616) was compared to heparin (n=1616) alone as medical management in patients with unstable angina/non-Q-wave myocardial infarction. In this study, the drug was started within 24 hours of the time the patient experienced chest pain. The mean age of the population was 62 years; 32% of the population was female and 25% had non-Q-wave myocardial infarction on presentation. Thirty percent had no ECG evidence of cardiac ischemia. Exclusion criteria were similar to PRISM-PLUS. The primary, prospectively identified endpoint was the composite endpoint of refractory ischemia, myocardial infarction or death after a 48-hour drug infusion with AGGRASTAT. The results are shown in Table 2.
| Composite Endpoint | AGGRASTAT (n=1616) |
Heparin (n=1616) |
Risk Reduction | p-value |
| 2 Days | 3.8% | 5.6% | 33% | 0.015 |
| 7 Days | 10.3% | 11.3% | 10% | 0.33 |
| 30 Days | 15.9% | 17.1% | 8% | 0.34 |
In the PRISM study, no adverse effect of AGGRASTAT on mortality at either 7 or 30 days was detected. This result is in conflict with the PRISM-PLUS study, where the arm that included AGGRASTAT without heparin (n=345) was dropped at an interim analysis by the Data Safety Monitoring Committee due to increased mortality at 7 days. A pooled analysis of the data from these two trials (PRISM and PRISM-PLUS) demonstrated that the effect of AGGRASTAT alone on mortality (at 7 and 30 days) was comparable to that of heparin alone.
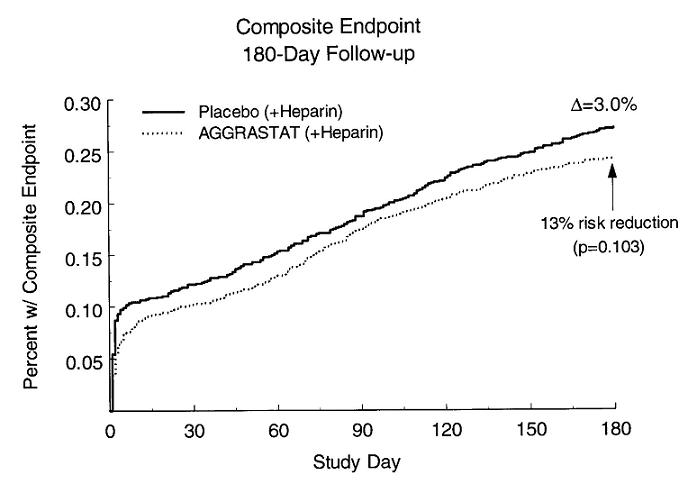
The RESTORE study (n=2141) was a randomized, controlled comparison of AGGRASTAT and placebo, each added to heparin, in patients undergoing PTCA or atherectomy within 72 hours of presentation with unstable angina or acute myocardial infarction. The mean age of the population was 59 years; 27% were female. Two-thirds of patients underwent angioplasty for unstable angina and the remainder in association with acute myocardial infarction. Exclusions included anatomy not amenable to angioplasty, contraindications to anticoagulation (see CONTRAINDICATIONS), platelet count <150,000/mm3, and creatinine >2.0 mg/dL. AGGRASTAT (with heparin) was initiated immediately prior to the angioplasty/atherectomy at a dose of 10 mcg/kg bolus (over 3 minutes) followed by an infusion of 0.15 mcg/kg/min along with a heparin bolus (bolus of 10,000 U, or 150 U/kg for patients <70 kg). The infusion dose of AGGRASTAT is 50% higher than the dose used in the PRISM-PLUS trial. AGGRASTAT was administered for a total of 36 hours. In general, heparin was to be discontinued at the conclusion of the angioplasty/atherectomy. Reasons for continued heparin included: imperfect outcome (e.g., large tear, intraluminal filling defect, or residual stenosis >40%), large thrombus load, continuing rest angina through the procedure, abrupt closure or very active artery during the procedure, or side branch occlusion. The primary endpoint was the composite of all deaths, non-fatal myocardial infarctions, and all repeat revascularization procedures at 30 days. For results see Table 3. A sub-study in RESTORE of angiograms after approximately 6 months found that AGGRASTAT had no significant effect on the extent of coronary artery restenosis following angioplasty.
| Composite Endpoint | AGGRASTAT (n=1071) |
Placebo (n=1070) |
Risk Reduction | p-value |
| 2 Days | 5.4% | 8.7% | 38% | 0.004 |
| 7 Days | 7.6% | 10.4% | 28% | 0.023 |
| 30 Days | 10.3% | 12.2% | 17% | 0.17 |
The risk reduction in the composite endpoint at 180 days is shown in the Kaplan-Meier curve below.
AGGRASTAT, in combination with heparin, is indicated for the treatment of acute coronary syndrome, including patients who are to be managed medically and those undergoing PTCA or atherectomy. In this setting, AGGRASTAT has been shown to decrease the rate of a combined endpoint of death, new myocardial infarction or refractory ischemia/repeat cardiac procedure (for discussion of trial results and for definition of acute coronary syndrome see CLINICAL PHARMACOLOGY, Clinical Trials).
AGGRASTAT has been studied in a setting, as described in Clinical Trials, that included aspirin and heparin.
AGGRASTAT is contraindicated in patients with:
Bleeding is the most common complication encountered during therapy with AGGRASTAT. Administration of AGGRASTAT is associated with an increase in bleeding events classified as both major and minor bleeding events by criteria developed by the Thrombolysis in Myocardial Infarction Study group (TIMI)². Most major bleeding associated with AGGRASTAT occurs at the arterial access site for cardiac catheterization. Fatal bleedings have been reported (see ADVERSE REACTIONS).
AGGRASTAT should be used with caution in patients with platelet count <150,000/mm3, in patients with hemorrhagic retinopathy, and in chronic hemodialysis patients.
Because AGGRASTAT inhibits platelet aggregation, caution should be employed when it is used with other drugs that affect hemostasis. The safety of AGGRASTAT when used in combination with thrombolytic agents has not been established.
During therapy with AGGRASTAT, patients should be monitored for potential bleeding. When bleeding cannot be controlled with pressure, infusion of AGGRASTAT and heparin should be discontinued.
Percutaneous Coronary Intervention — Care of the femoral artery access site: Therapy with AGGRASTAT is associated with increases in bleeding rates particularly at the site of arterial access for femoral sheath placement. Care should be taken when attempting vascular access that only the anterior wall of the femoral artery is punctured. Prior to pulling the sheath, heparin should be discontinued for 3-4 hours and activated clotting time (ACT) <180 seconds or APTT <45 seconds should be documented. Care should be taken to obtain proper hemostasis after removal of the sheaths using standard compressive techniques followed by close observation. While the vascular sheath is in place, patients should be maintained on complete bed rest with the head of the bed elevated 30° and the affected limb restrained in a straight position. Sheath hemostasis should be achieved at least 4 hours before hospital discharge.
Minimize Vascular and Other Trauma: Other arterial and venous punctures, epidural procedures, intramuscular injections, and the use of urinary catheters, nasotracheal intubation and nasogastric tubes should be minimized. When obtaining intravenous access, non-compressible sites (e.g., subclavian or jugular veins) should be avoided.
Laboratory Monitoring: Platelet counts, and hemoglobin and hematocrit should be monitored prior to treatment, within 6 hours following the loading infusion, and at least daily thereafter during therapy with AGGRASTAT (or more frequently if there is evidence of significant decline). In patients who have previously received GP IIb/IIIa receptor antagonists, consideration should be given to earlier monitoring of platelet count. If the patient experiences a platelet decrease to <90,000/mm3, additional platelet counts should be performed to exclude pseudothrombocytopenia. If thrombocytopenia is confirmed, AGGRASTAT and heparin should be discontinued and the condition appropriately monitored and treated.
In addition, the activated partial thromboplastin time (APTT) should be determined before treatment and the anticoagulant effects of heparin should be carefully monitored by repeated determinations of APTT and the dose should be adjusted accordingly (see also DOSAGE AND ADMINISTRATION). Potentially life-threatening bleeding may occur especially when heparin is administered with other products affecting hemostasis, such as GP IIb/IIIa receptor antagonists. To monitor unfractionated heparin, APTT should be monitored 6 hours after the start of the heparin infusion; heparin should be adjusted to maintain APTT at approximately 2 times control.
In clinical studies, patients with severe renal insufficiency (creatinine clearance <30 mL/min) showed decreased plasma clearance of AGGRASTAT. The dosage of AGGRASTAT should be reduced in these patients (see DOSAGE AND ADMINISTRATION and CLINICAL PHARMACOLOGY, Clinical Trials).
AGGRASTAT has been studied on a background of aspirin and heparin.
The use of AGGRASTAT, in combination with heparin and aspirin, has been associated with an increase in bleeding compared to heparin and aspirin alone (see ADVERSE REACTIONS). Caution should be employed when AGGRASTAT is used with other drugs that affect hemostasis (e.g., warfarin). No information is available about the concomitant use of AGGRASTAT with thrombolytic agents (see PRECAUTIONS, Bleeding Precautions).
In a sub-set of patients (n=762) in the PRISM study, the plasma clearance of tirofiban in patients receiving one of the following drugs was compared to that in patients not receiving that drug. There were no clinically significant effects of co-administration of these drugs on the plasma clearance of tirofiban: acebutolol, acetaminophen, alprazolam, amlodipine, aspirin preparations, atenolol, bromazepam, captopril, diazepam, digoxin, diltiazem, docusate sodium, enalapril, furosemide, glyburide, heparin, insulin, isosorbide, lorazepam, lovastatin, metoclopramide, metoprolol, morphine, nifedipine, nitrate preparations, oxazepam, potassium chloride, propranolol, ranitidine, simvastatin, sucralfate and temazepam. Patients who received levothyroxine or omeprazole along with AGGRASTAT had a higher rate of clearance of AGGRASTAT. The clinical significance of this is unknown.
The carcinogenic potential of AGGRASTAT has not been evaluated.
Tirofiban HCI was negative in the in vitro microbial mutagenesis and V-79 mammalian cell mutagenesis assays. In addition, there was no evidence of direct genotoxicity in the in vitro alkaline elution and in vitro chromosomal aberration assays. There was no induction of chromosomal aberrations in bone marrow cells of male mice after the administration of intravenous doses up to 5 mg tirofiban/kg (about 3 times the maximum recommended daily human dose when compared on a body surface area basis).
Fertility and reproductive performance were not affected in studies with male and female rats given intravenous doses of tirofiban hydrochloride up to 5 mg/kg/day (about 5 times the maximum recommended daily human dose when compared on a body surface area basis).
Pregnancy Category B
Tirofiban has been shown to cross the placenta in pregnant rats and rabbits. Studies with tirofiban HCI at intravenous doses up to 5 mg/kg/day (about 5 and 13 times the maximum recommended daily human dose for rat and rabbit, respectively, when compared on a body surface area basis) have revealed no harm to the fetus. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
It is not known whether tirofiban is excreted in human milk. However, significant levels of tirofiban were shown to be present in rat milk. Because many drugs are excreted in human milk, and because of the potential for adverse effects on the nursing infant, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
Safety and effectiveness of AGGRASTAT in pediatric patients (<18 years old) have not been established.
Of the total number of patients in controlled clinical studies of AGGRASTAT, 42.8% were 65 years and over, while 11.7% were 75 and over. With respect to efficacy, the effect of AGGRASTAT in the elderly (≥65 years) appeared similar to that seen in younger patients (<65 years). Elderly patients receiving AGGRASTAT with heparin or heparin alone had a higher incidence of bleeding complications than younger patients, but the incremental risk of bleeding in patients treated with AGGRASTAT in combination with heparin compared to the risk in patients treated with heparin alone was similar regardless of age. The overall incidence of non-bleeding adverse events was higher in older patients (compared to younger patients) but this was true both for AGGRASTAT with heparin and heparin alone. No dose adjustment is recommended for the elderly population (see DOSAGE AND ADMINISTRATION, Recommended Dosage).
In clinical trials, 1946 patients received AGGRASTAT in combination with heparin and 2002 patients received AGGRASTAT alone. Duration of exposure was up to 116 hours. 43% of the population was >65 years of age and approximately 30% of patients were female.
The most common drug-related adverse event reported during therapy with AGGRASTAT when used concomitantly with heparin and aspirin, was bleeding (usually reported by the investigators as oozing or mild). The incidences of major and minor bleeding using the TIMI criteria in the PRISM-PLUS and RESTORE studies are shown below.
| ||||
| PRISM-PLUS* (UAP/Non-Q-Wave MI Study) |
RESTORE* (Angioplasty/Atherectomy Study) |
|||
| Bleeding | AGGRASTAT† + Heparin‡ (n=773) %(n) |
Heparin‡ (n=797) %(n) |
AGGRASTAT§ + Heparin ¶ (n=1071) %(n) |
Heparin¶ (n=1070) %(n) |
| Major Bleeding (TIMI Criteria)# | 1.4 (11) | 0.8 (6) | 2.2 (24) | 1.6 (17) |
| Minor Bleeding (TIMI Criteria)Þ | 10.5 (81) | 8.0 (64) | 12.0 (129) | 6.3 (67) |
| Transfusions | 4.0 (31) | 2.8 (22) | 4.3 (46) | 2.5 (27) |
There were no reports of intracranial bleeding in the PRISM-PLUS study for AGGRASTAT in combination with heparin or in the heparin control group. The incidence of intracranial bleeding in the RESTORE study was 0.1% for AGGRASTAT in combination with heparin and 0.3% for the control group (which received heparin). In the PRISM-PLUS study, the incidences of retroperitoneal bleeding reported for AGGRASTAT in combination with heparin, and for the heparin control group were 0.0% and 0.1%, respectively. In the RESTORE study, the incidences of retroperitoneal bleeding reported for AGGRASTAT in combination with heparin, and the control group were 0.6% and 0.3%, respectively. The incidences of TIMI major gastrointestinal and genitourinary bleeding for AGGRASTAT in combination with heparin in the PRISM-PLUS study were 0.1% and 0.1%, respectively; the incidences in the RESTORE study for AGGRASTAT in combination with heparin were 0.2% and 0.0%, respectively.
The incidence rates of TIMI major bleeding in patients undergoing percutaneous procedures in PRISM-PLUS are shown below.
| AGGRASTAT + Heparin | Heparin | |||
| n | % | n | % | |
| Prior to Procedures | 2/773 | 0.3 | 1/797 | 0.1 |
| Following Angiography | 9/697 | 1.3 | 5/708 | 0.7 |
| Following PTCA | 6/239 | 2.5 | 5/236 | 2.2 |
The incidence rates of TIMI major bleeding (in some cases possibly reflecting hemodilution rather than actual bleeding) in patients undergoing CABG in the PRISM-PLUS and RESTORE studies within one day of discontinuation of AGGRASTAT are shown below.
| AGGRASTAT +Heparin | Heparin | |||
| n | % | n | % | |
| PRISM-PLUS | 5/29 | 17.2 | 11/31 | 35.4 |
| RESTORE | 3/12 | 25.0 | 6/16 | 37.5 |
Female patients and elderly patients receiving AGGRASTAT with heparin or heparin alone had a higher incidence of bleeding complications than male patients or younger patients. The incremental risk of bleeding in patients treated with AGGRASTAT in combination with heparin over the risk in patients treated with heparin alone was comparable regardless of age or gender. No dose adjustment is recommended for these populations (see DOSAGE AND ADMINISTRATION, Recommended Dosage).
The incidences of non-bleeding adverse events that occurred at an incidence of >1% and numerically higher than control, regardless of drug relationship, are shown below:
| AGGRASTAT + Heparin (n=1953) % |
Heparin (n=1887) % |
|
| Body as a Whole | ||
| Edema/swelling | 2 | 1 |
| Pain, pelvic | 6 | 5 |
| Reaction, vasovagal | 2 | 1 |
| Cardiovascular System | ||
| Bradycardia | 4 | 3 |
| Dissection, coronary artery | 5 | 4 |
| Musculoskeletal System | ||
| Pain, leg | 3 | 2 |
| Nervous System/Psychiatric | ||
| Dizziness | 3 | 2 |
| Skin and Skin Appendage | ||
| Sweating | 2 | 1 |
Other non-bleeding side effects (considered at least possibly related to treatment) reported at a >1% rate with AGGRASTAT administered concomitantly with heparin were nausea, fever, and headache; these side effects were reported at a similar rate in the heparin group.
In clinical studies, the incidences of adverse events were generally similar among different races, patients with or without hypertension, patients with or without diabetes mellitus, and patients with or without hypercholesteremia.
The overall incidence of non-bleeding adverse events was higher in female patients (compared to male patients) and older patients (compared to younger patients). However, the incidences of non-bleeding adverse events in these patients were comparable between the AGGRASTAT with heparin and the heparin alone groups. (See above for bleeding adverse events.)
Although no patients in the clinical trial database developed anaphylaxis and/or hives requiring discontinuation of the infusion of tirofiban, anaphylaxis has been reported in post-marketing experience (see also Post-Marketing Experience, Hypersensitivity). No information is available regarding the development of antibodies to tirofiban.
The most frequently observed laboratory adverse events in patients receiving AGGRASTAT concomitantly with heparin were related to bleeding. Decreases in hemoglobin (2.1%) and hematocrit (2.2%) were observed in the group receiving AGGRASTAT compared to 3.1% and 2.6%, respectively, in the heparin group. Increases in the presence of urine and fecal occult blood were also observed (10.7% and 18.3%, respectively) in the group receiving AGGRASTAT compared to 7.8% and 12.2%, respectively, in the heparin group.
Patients treated with AGGRASTAT, with heparin, were more likely to experience decreases in platelet counts than the control group. These decreases were reversible upon discontinuation of AGGRASTAT. The percentage of patients with a decrease of platelets to <90,000/mm3 was 1.5%, compared with 0.6% in the patients who received heparin alone. The percentage of patients with a decrease of platelets to <50,000/mm3 was 0.3%, compared with 0.1% of the patients who received heparin alone. Platelet decreases have been observed in patients with no prior history of thrombocytopenia upon readministration of GP IIb/IIIa receptor antagonists.
The following additional adverse reactions have been reported in post-marketing experience: Bleeding: Intracranial bleeding, retroperitoneal bleeding, hemopericardium, pulmonary (alveolar) hemorrhage, and spinal-epidural hematoma. Fatal bleeding events have been reported;Body as a Whole: Acute and/or severe decreases in platelet counts which may be associated with chills, low-grade fever, or bleeding complications (see Laboratory Findings above);Hypersensitivity: Severe allergic reactions including anaphylactic reactions. The reported cases have occurred during the first day of tirofiban infusion, during initial treatment, and during readministration of tirofiban. Some cases have been associated with severe thrombocytopenia (platelet counts <10,000/mm3).
In clinical trials, inadvertent overdosage with AGGRASTAT occurred in doses up to 5 times and 2 times the recommended dose for bolus administration and loading infusion, respectively. Inadvertent overdosage occurred in doses up to 9.8 times the 0.15 mcg/kg/min maintenance infusion rate.
The most frequently reported manifestation of overdosage was bleeding, primarily minor mucocutaneous bleeding events and minor bleeding at the sites of cardiac catheterization (see PRECAUTIONS, Bleeding Precautions).
Overdosage of AGGRASTAT should be treated by assessment of the patient’s clinical condition and cessation or adjustment of the drug infusion as appropriate.
AGGRASTAT can be removed by hemodialysis.
In the clinical studies, patients received aspirin, unless it was contraindicated, and heparin. AGGRASTAT and heparin can be administered through the same intravenous catheter.
AGGRASTAT is intended for intravenous delivery using sterile equipment and technique. Do not add other drugs or remove solution directly from the bag with a syringe. Do not use plastic containers in series connections; such use can result in air embolism by drawing air from the first container if it is empty of solution. Any unused solution should be discarded.
AGGRASTAT Injection Premixed is supplied as 100 mL or 250 mL of 0.9% sodium chloride containing 50 mcg/mL tirofiban. It is supplied in lNTRAVIA³ containers (PL 2408 plastic). To open the lNTRAVIA container, first tear off its foil overpouch. The plastic may be somewhat opaque because of moisture absorption during sterilization; the opacity will diminish gradually. Check for leaks by squeezing the inner bag firmly; if any leaks are found, the sterility is suspect and the solution should be discarded. Do not use unless the solution is clear and the seal is intact. Suspend the container from its eyelet support, remove the plastic protector from the outlet port, and attach a conventional administration set.
AGGRASTAT may be administered in the same intravenous line as atropine sulfate, dobutamine, dopamine, epinephrine HCl, famotidine injection, furosemide, lidocaine, midazolam HCl, morphine sulfate, nitroglycerin, potassium chloride, and propranolol HCl. AGGRASTAT should not be administered in the same intravenous line as diazepam.
In most patients, AGGRASTAT should be administered intravenously, at an initial rate of 0.4 mcg/kg/min for 30 minutes and then continued at 0.1 mcg/kg/min.
Patients with severe renal insufficiency (creatinine clearance <30 mL/min) should receive half the usual rate of infusion (see PRECAUTIONS, Severe Renal Insufficiency and CLINICAL PHARMACOLOGY, Pharmacokinetics, Special Populations, Renal Insufficiency).The table below is provided as a guide to dosage adjustment by weight.
| Patient Weight (kg) | Most Patients | Severe Renal Impairment | ||
| 30 Min Loading Infusion Rate (mL/hr) | Maintenance Infusion Rate (mL/hr) | 30 Min Loading Infusion Rate (mL/hr) | Maintenance Infusion Rate (mL/hr) | |
| 30-37 | 16 | 4 | 8 | 2 |
| 38-45 | 20 | 5 | 10 | 3 |
| 46-54 | 24 | 6 | 12 | 3 |
| 55-62 | 28 | 7 | 14 | 4 |
| 63-70 | 32 | 8 | 16 | 4 |
| 71-79 | 36 | 9 | 18 | 5 |
| 80-87 | 40 | 10 | 20 | 5 |
| 88-95 | 44 | 11 | 22 | 6 |
| 96-104 | 48 | 12 | 24 | 6 |
| 105-112 | 52 | 13 | 26 | 7 |
| 113-120 | 56 | 14 | 28 | 7 |
| 121-128 | 60 | 15 | 30 | 8 |
| 129-137 | 64 | 16 | 32 | 8 |
| 138-145 | 68 | 17 | 34 | 9 |
| 146-153 | 72 | 18 | 36 | 9 |
No dosage adjustment is recommended for elderly or female patients (see PRECAUTIONS, Geriatric Use). In PRISM-PLUS, AGGRASTAT was administered in combination with heparin for 48 to 108 hours. The infusion should be continued through angiography and for 12 to 24 hours after angioplasty or atherectomy.
FOR INTRAVENOUS USE ONLY
Rx Only
AGGRASTAT Injection Premixed 5 mg tirofiban per 100 mL (50 mcg per mL) and 12.5 mg tirofiban per 250 mL (50 mcg per mL) are clear, non-preserved, sterile solutions premixed in a vehicle made iso-osmotic with sodium chloride, and are supplied as follows:
NDC 25208-002-01, 100 mL single-dose lNTRAVIA containers (PL 2408 Plastic).
NDC 25208-002-02, 250 mL single-dose lNTRAVIA containers (PL 2408 Plastic).
Storage
AGGRASTAT Injection Premixed
Store at 25°C (77°F) with excursions permitted between 15-30°C (59-86°F) (see USP Controlled Room Temperature). Do not freeze. Protect from light during storage.
Patent: www.medicure.com/aggrastat/patents
AGGRASTAT (Tirofiban Hydrochloride Injection Premixed) is manufactured for:
MEDICURE INTERNATIONAL, INC.
by:
BAXTER HEALTHCARE CORPORATION
Deerfield, IL 60015, USA
Distributed by:
MEDICURE PHARMA, INC.
Somerset, NJ 08873 USA
1-800-509-0544
Issued April 2012
Printed in USA
¹ Registered trademark of Medicure International Inc.
© 1998 Copyright used under license.
All rights reserved.
² Bovill, E.G.; et al.: Hemorrhagic Events during Therapy with Recombinant Tissue-Type Plasminogen Activator, Heparin, and Aspirin for Acute Myocardial Infarction, Results of the Thrombolysis in Myocardial Infarction (TIMI) Phase II Trial, Annals of Internal Medicine, 115(4): 256-265, 1991.
³ Intravia is a registered trademark of Baxter International Inc.
Insert

Container

Carton

|
AGGRASTAT
tirofiban injection, solution | ||||||||||||||||||||||||
| ||||||||||||||||||||||||
| ||||||||||||||||||||||||
| ||||||||||||||||||||||||
| ||||||||||||||||||||||||
| ||||||||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA020913 | 05/14/1998 | |
| Labeler - Medicure International Inc (860240324) |
| Registrant - Medicure International Inc (860240324) |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Baxter Healthcare Corporation | 059140764 | manufacture, analysis, pack, label | |
